Molecular Docking Task
Molecular docking plays a crucial role in drug discovery, virtual screening, identification of potential lead compounds by estimating binding strength and orientation within the active site. Herein this task enables the intraction study of small molecules (ligands) to a target protein using molecular docking method integrated into the platform for ease of use, fast binding energy calculation along with ligand pose calculations inside the binding pockets based on energy affinity in a fully automated pipeline.
Overview
The docking interface allows users to:
Upload a prepared protein structure (.pdbqt format)
Upload a ligand structure (.pdbqt)
Define the docking box interactively or by coordinate entry
Run molecular docking directly on the server
View/download docked poses and binding scores
Input Requirements
The following fields must be filled before submitting a docking task:
Upload a protein structure in .pdbqt format prepared via the platform’s Protein Preparation module. Before uploding the protein.pdbqt file user has to ensure the file has no waters, heteroatoms, or additional chains.
Upload a ligand structure in .pdbqt format generated using the Ligand Preparation module or external tools. User has to make sure torsions and charges are properly assigned.
The box center defines the central point (X, Y, Z coordinates) of the docking search space defining the box center with X, Y, Z coordinates to ensure the docking box is aligned with the binding pocket or active site of the receptor protein.
Defining the box size in molecular docking is important because it sets the search space—the 3D region where the docking algorithm will look for potential binding poses of ligand in the protein.
Here in this module we are using grid-based scoring function. larger box means more grid points, which significantly increases memory and CPU time.
Exhaustiveness is a parameter that defines the amount of computational effort spent searching for optimal docking poses. Higher values result in more extensive searches, potentially yielding better results but requiring longer run times.(default = 8).
Optional. Number of binding poses to generate (default = 9).
Warning
Ensure the box covers the full binding pocket. Too small a box may lead to inaccurate or missed interactions.
How to Use the Interface
+--------------------------+
| Start |
+--------------------------+
|
v
+----------------+ +---------------------+
| Upload Receptor | | Upload Ligand(s) |
| (receptor.pdbqt)| | (ligand.pdbqt/.zip) |
+-----------------+ +---------------------+
\ /
\ /
v v
+----------------------+
| Set Docking Parameters|
| Center, Size, etc. |
+----------------------+
|
v
+--------------------------+
| Click 'Run Docking Task'|
+--------------------------+
|
v
+--------------------------+
| View in Job History |
+--------------------------+
|
v
+--------------------------+
| Docking Results Ready |
+--------------------------+
Warning
Size of X, Y, Z should not exceed 30, 30, 30 Å.
Output
The Molecular Docking Results section provides an interactive visualization of docking poses along with relevant metadata and downloadable result files.
Docking Pose Selection
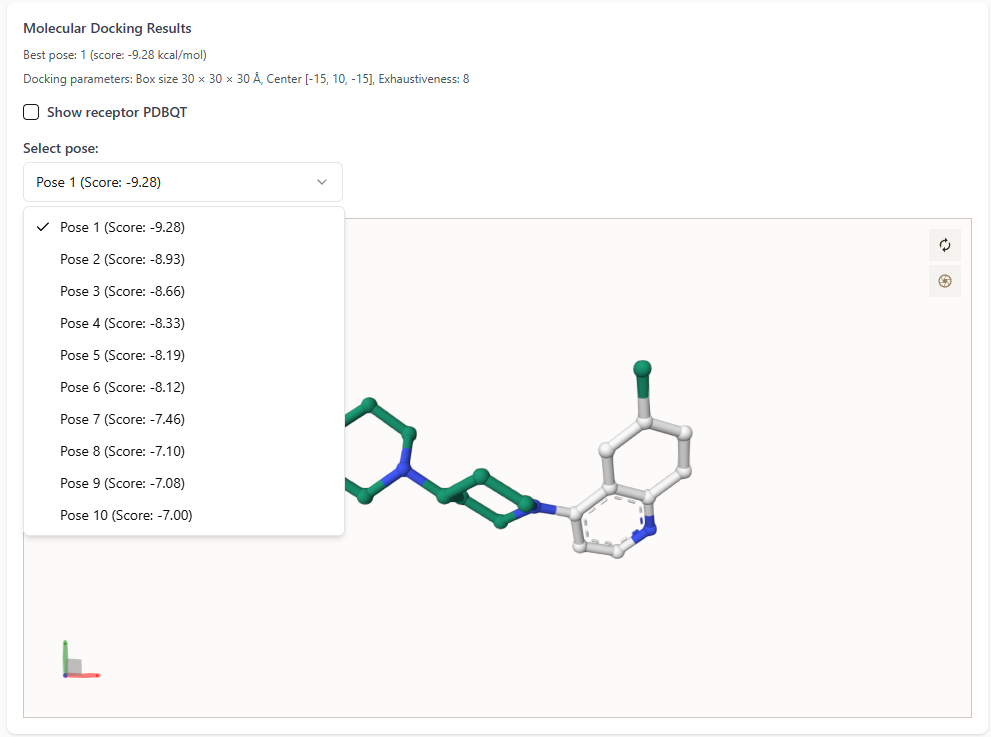
On the left-hand side of the docking results interface:
The best docking pose is highlighted with its score (in kcal/mol).
- Users can explore all docking poses by clicking the dropdown menu.
Each pose is listed along with its corresponding docking score for comparison.
This enables users to visually inspect and analyze multiple docking conformations with ease.
Ligand-Receptor Interaction Visualization
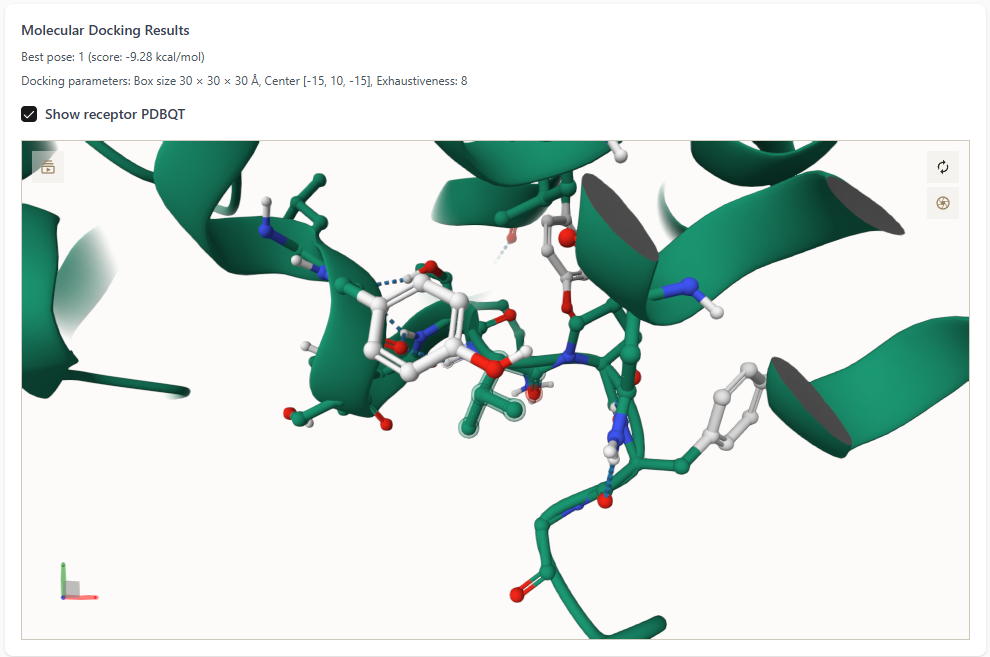
Upon selecting the “Show receptor PDBQT” checkbox:
The protein receptor is displayed in the 3D viewer.
The best ligand pose is shown docked inside the protein’s binding pocket.
- This detailed view helps users assess the quality of interactions,
such as hydrogen bonding, hydrophobic contacts, and spatial fit.
Metadata and Other files
On the right-hand side, a metadata box provides:
Docking parameters: box size, center coordinates, and exhaustiveness.
Pose details: total number of poses and their respective scores.
Additional molecular properties if available.
Two result files are available for download:
`binding_energy.csv` — contains docking scores for all poses.
`ligands_pose.txt` — contains atomic coordinates for each pose in PDBQT format.
These outputs support downstream analysis or integration into pipelines.
Warning
Do not upload raw .pdb files. Always convert them to .pdbqt using platform modules.
Note
The platform currently assumes a rigid receptor and excludes water molecules during docking.
More negative binding energy suggests better affinity, but verify visually.
Best Practices
Visually inspect binding poses
Confirm docking box accuracy using known ligands
Run multiple docking simulations to account for ligand flexibility
Future Enhancements
Flexible receptor support
Auto-detection of binding pockets
Post-docking energy minimization